Welcome to the Michael B. Hall Research Group
Our group applies "state-of-the-art" theoretical techniques to chemical problems of experimental interest in inorganic, organometallic, biological, and materials chemistry.
Our underutilized reserves of methane have provided the impetus for many research groups to study C-H bond activation. Detailed kinetics of the early stages of the C-H bonds activated by a tris-pyrazolylborate(Tp) rhodium carbonyl show the formation of two successive intermediates. Our preliminary calculations have identified these as a weakly solvated trihapto-Tp followed by a more strongly solvated dihapto-Tp complex, an interpretation that is quite different from the original ones suggested in the experimental work. Recent work has explained the unexpected dramatic change in C-H activation rates for cyclohexane vs. cycloheptane.
Water splitting and the reduction of protons to hydrogen continues to be a major interest, particularly the mechanism for Hydrogenases and related model complexes. Recent work has shown how the electro storage capacity of the nitrosyl ligand moderates the mechanism for electrochemical hydrogen production.
In addition to the nitrosyl ligand, other non-innocent ligands supported by transition metals play key roles in many catalytic transformations. Recently, we have shown that the XYZ ligand supported by ruthenium transforms methanol to carbon dioxide and hydrogen with a mechanism that involves proton and hydride transfers to the ligand framework rather than the metal. Thus, the transition metal plays the ‘spectator’ role in this system.
-
 Hall Research Group
Hall Research Group -
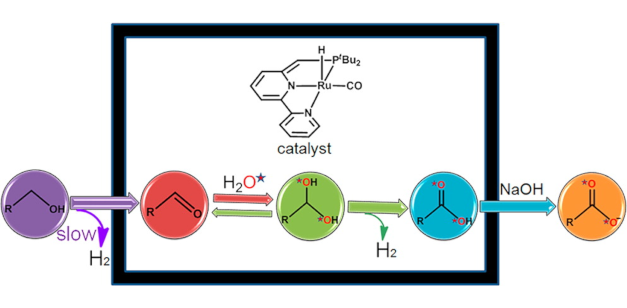 Mechanism of the Formation of Carboxylate from Alcohols and Water Catalyzed by a Bipyridine-Based Ruthenium Complex: A Computational Study
Mechanism of the Formation of Carboxylate from Alcohols and Water Catalyzed by a Bipyridine-Based Ruthenium Complex: A Computational StudyDOI: 10.1021/ja410541v
-
![JACS abstract figure for Intramolecular Iron-Mediated C–H Bond Heterolysis with an Assist of Pendant Base in a [FeFe]-Hydrogenase Model publication](images/6.png) Intramolecular Iron-Mediated C–H Bond Heterolysis with an Assist of Pendant Base in a [FeFe]-Hydrogenase Model
Intramolecular Iron-Mediated C–H Bond Heterolysis with an Assist of Pendant Base in a [FeFe]-Hydrogenase ModelDOI: 10.1021/ja5078014
-
![JACS abstract figure for Biomimetics of [NiFe]-Hydrogenase: Nickel- or Iron-Centered Proton Reduction Catalysis? publication](images/5.png) Biomimetics of [NiFe]-Hydrogenase: Nickel- or Iron-Centered Proton Reduction Catalysis?
Biomimetics of [NiFe]-Hydrogenase: Nickel- or Iron-Centered Proton Reduction Catalysis?DOI: 10.1021/jacs.7b10425
-
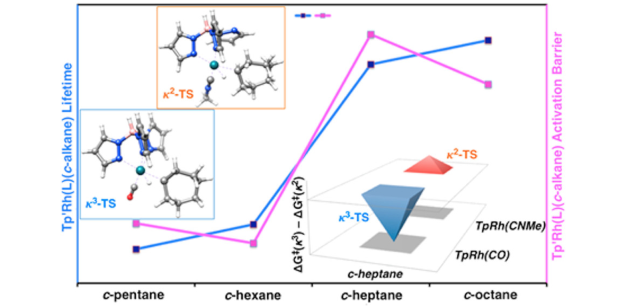 Probing the Carbon–Hydrogen Activation of Alkanes Following Photolysis of Tp′Rh(CNR)(carbodiimide): A Computational and Time-Resolved Infrared Spectroscopic Study
Probing the Carbon–Hydrogen Activation of Alkanes Following Photolysis of Tp′Rh(CNR)(carbodiimide): A Computational and Time-Resolved Infrared Spectroscopic StudyDOI: 10.1021/jacs.7b12152
-
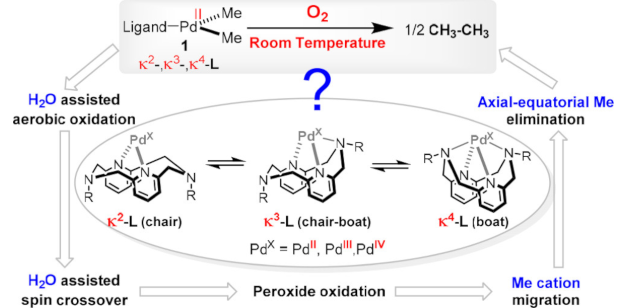 Unraveling the Role of a Flexible Tetradentate Ligand in the Aerobic Oxidative Carbon–Carbon Bond Formation with Palladium Complexes: A Computational Mechanistic Study
Unraveling the Role of a Flexible Tetradentate Ligand in the Aerobic Oxidative Carbon–Carbon Bond Formation with Palladium Complexes: A Computational Mechanistic StudyDOI: 10.1021/jacs.7b11701