
~ M. Y. DARENSBOURG ~
RESEARCH LABORATORIES
~ M. Y. DARENSBOURG ~
RESEARCH LABORATORIES
|
Recent Publications |
|
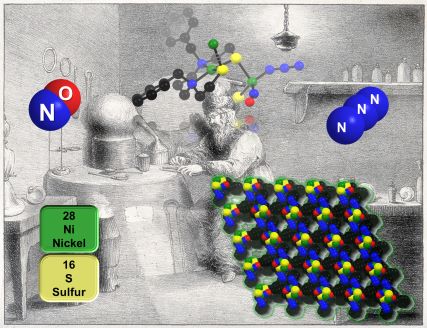 A sulfur-templated Ni–Ni′ coordination polymer that relies on a polarizable nickel nitrosyl hub. Dalton Trans.,, 2025 DOI:https://doi.org/10.1039/D4DT03174A |
The templating properties of a diaza-nickel-cis-dithiolate towards triphenylphosphine gold(I), yielding a transoid [Ni(N2S2)·2Au(PPh3)] complex (T. A. Pinder, S. K. Montalvo, A. M. Lunsford, C.-H. Hsieh, J. H. Reibenspies and M. Y. Darensbourg, Dalton Trans., 2014, 43, 138–144) suggested that a suitable analogue of d10-Au(I), i.e., {Ni(NO)}10, could generate a tetrahedral nickel node for a [Ni(N2S2)·2Ni(NO)(X)]n coordination polymer. Monomeric precursors, derived from Feltham's [(Ph3P)2Ni(NO)(Cl)] (R. D. Feltham, Inorg. Chem., 1964, 3, 116–119) produced the bidentate/sulfur-chelated [Ni(N2S2)·Ni(NO)(X)] species with loss of PPh3. Exchange of Cl− by azide, N3−, in the {Ni(NO)}10 synthon led to the balance of electrophilicity at Ni(NO) and non-covalent (H-bonding and van der Waals) interactions that stabilized the extended chain of bridging sulfurs, in transoid connectivities, between a square planar NiII and a tetrahedral Ni, the latter within the electronic and spin-delocalized {Ni(NO)}10 system. This study defines a new path that creates coordination polymers using metallodithiolates, the success of which, in this case, depends on the highly polarizable {Ni(NO)}10 unit. |
 Nitric oxide transfer between nominal Fe and Co biomimetics of the nitrile hydratase active site Biological Inorg. Chem., 2025 DOI:https://doi.org/10.1007/s00775-024-02092-8 |
Related to the inactive form of nitrile hydratase, NHase, that contains Fe(NO) within tripeptide N<2S2 binding environment, the NO transfer reactivity of (bis-mercaptoethane diazacycloheptane)Fe(NO) and (bis-mercaptoethane diazadimethylethane)Fe(NO) is compared to Co(NO) analogs. Acceptors of NO include cobalt octaethylporphyrin and the [(N2S2)M] dimeric precursors in the synthesis of the Fe(NO) and Co(NO) biomimetics. Qualitative rates are augmented by a definitive kinetic study finding that rates of NO transfer from (N2S2)M(NO) to [(N2S2)M′]2 are dependent on M and M′ as well as the hydrocarbon N to N and N to S linkers. We conclude that while Fe(NO) and Co(NO) units are similar in chemical stability, minor first coordination sphere differences may favor the former, Fe(NO), consistent with the discovery of Fe(NO), but not Co(NO), in the as-isolated NHase active site. |
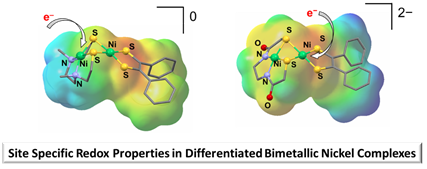 Site specific redox properties in ligand differentiated di-nickel complexes inspired by the acetyl CoA synthase active site. Dalton Trans., 2024, Advanced Article, (Collaboration with reMIND) DOI: https://doi.org/10.1039/D4DT00306C |
These complexes, comprised of Ni(N2S2)-Ni(dithiolene) S-bridged units, serve as a platform to interrogate the positions of added electrons. Tuning of the ligand substituents controls electron uptake in S-bridged dinickel complexes. |
 Development of (NO)Fe(N2S2) as a Metallodithiolate Spin Probe Ligand: A Case Study Approach ACC. Chem. Res 2024, 57, 6, 831–844, (Collaboration with reMIND) DOI: 10.1021/acs.accounts.3c00667 |
Common motifs for biocatalysts developed in evolutionary biology include the placement of metals in proximity with flexible sulfur bridges, as well as the presence of π-acidic/delocalizing ligands. This Account delves into the development of an (NO)Fe(N2S2) metallodithiolate ligand that harnesses these principles. The Fe(NO) unit is the centroid of an N2S2 donor field which as a whole is capable of serving as a redox-active, bidentate S-donor ligand. Its paramagnetism as well as the v(NO) vibrational monitor can be exploited in development of new classes of heterobimetallic complexes. Four examples in which the unpaired electron on the {Fe(NO)}7 unit is spin paired with adjacent or distant paramagnets find a range of magnetic coupling between Fe(NO) from −3 to −1200 cm−1. |
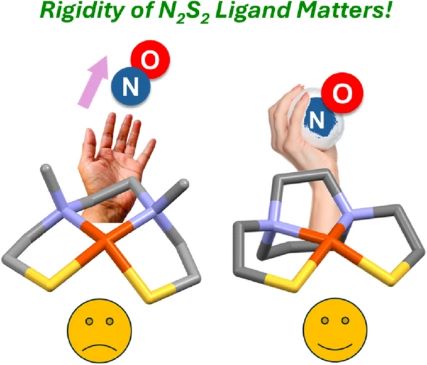
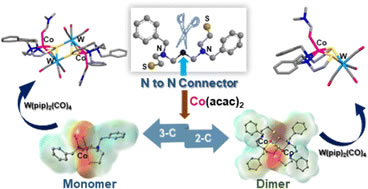 A single carbon atom controls the geometry and reactivity of CoII(N2S2) complexes Chem. Commun., 2024, 60, 1128-1131 DOI: https://doi.org/10.1039/D3CC05394F |
Three- vs. two-carbon N-to-N connectors give rise to monomeric, tetrahedral N2S2Co(II) (μeff = 4.24 BM) or dimeric [(N2S2)Co(II)]2 (diamagnetic) complexes, respectively. Differences in the derivative products of the Lewis acid receivers, W0(CO)3 and W0(CO)4, illustrate nucleophilicity of the thiolate sulfur lone pairs in each case, as well as their structural control. |
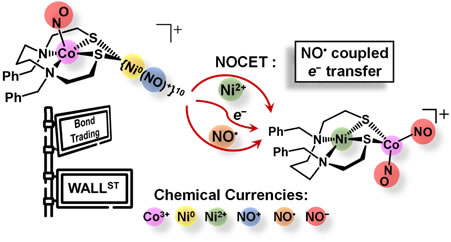 Bond Trading: Intramolecular Metal and Ligand Exchange within a NO/Ni/Co Complex Advanced Science 2023, 11, 2307113 DOI: 10.1002/advs.202307113 |
With the goal of generating hetero-redox levels on metals as well as on nitric oxide (NO), metallodithiolate (N2S2)CoIII(NO−), N2S2 = N,N- dibenzyl-3,7-diazanonane-1,9-dithiolate, is introduced as ligand to a well-characterized labile [Ni0(NO)+] synthon. The reaction between [Ni0(NO+)] and [CoIII(NO−)] has led to a remarkable electronic and ligand redistribution to form a heterobimetallic dinitrosyl cobalt [(N2S2)NiII∙Co(NO)2]+ complex with formal two electron oxidation state switches concomitant with the nickel extraction or transfer as NiII into the N2S2 ligand binding site. To date, this is the first reported heterobimetallic cobalt dinitrosyl complex. |
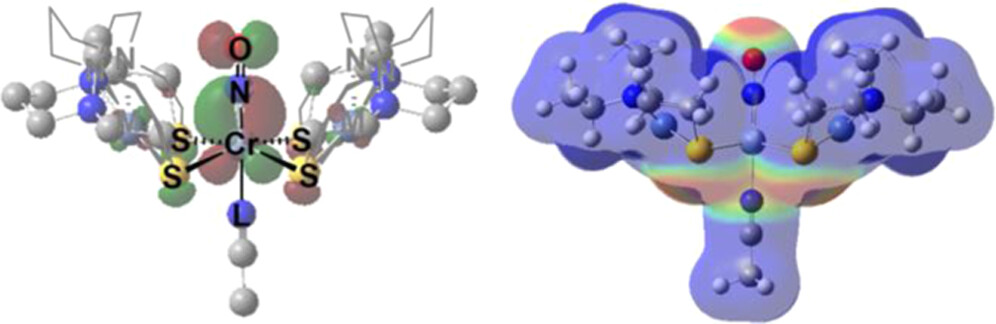 Sulfur Lone Pairs Control Topology in Heterotrimetallic Complexes: An Experimental and Theoretical Study ACS Org. Inorg. Au 2023, 3, 6, 393-402. (Collaboration with reMIND) DOI: https://doi.org/10.1021/acsorginorgau.3c00025 |
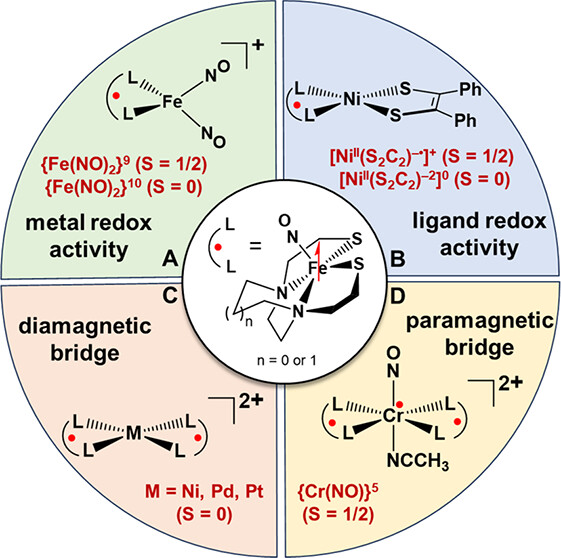
Heterotrimetallic complexes with (N2S2)M metallodithiolates, M = Ni2+, [Fe(NO)]2+, and [Co(NO)]2+, as bidentate chelating ligands to a central trans-Cr(NO)(MeCN) unit were characterized as the first members of a new class, NiCrNi, FeCrFe, CoCrCo. The complexes exhibit a cisoid structural topology, ascribed to the stereoactivity of the available lone pair(s) on the sulfur donors, resulting in a dispersed, electropositive pocket from the N/N and N/S hydrocarbon linkers wherein the Cr-NO site is housed. Computational studies explored alternative isomers (transoid and inverted cisoid) that suggest a combination of electronic and steric effects govern the geometrical selectivity. Electrostatic potential maps readily display the dominant electronegative potential from the sulfurs which force the NO to the electropositive pocket. The available S lone pairs work in synergy with the π-withdrawing ability of NO to lift Cr out of the S4 plane toward the NO and stabilize the geometry. The metallodithiolate ligands bound to Cr(NO) thus find structural consistency across the three congeners. Although the dinitrosyl [(bme-dach)Co(NO)-Mo(NO)(MeCN)-(bme-dach)Co(MeCN)][PF6]2 (CoMoCo′) analogue displays chemical noninnocence and a partial Mo–Co bond toward (N2S2)Co′(NCCH3) in an “asymmetric butterfly” topology [Guerrero-Almaraz, P. Inorg. Chem. 2021, 60(21), 15975–15979], the stability of the {Cr(NO)}5 unit prohibits such bond rearrangement. Magnetism and EPR studies illustrate spin coupling across the sulfur thiolate sulfur bridges. |
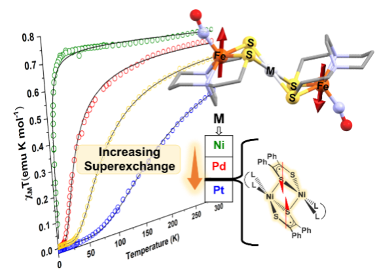 Magnetic coupling between Fe(NO) spin probe ligands through diamagnetic NiII, PdII and PtII tetrathiolate bridges Chem. Sci. 2023, 14, 9167-9174. (Collaboration with reMIND) DOI: https://doi.org/10.1039/D3SC01546G |
Reaction of the nitrosylated-iron metallodithiolate ligand, paramagnetic (NO)Fe(N2S2), with [M(CH3CN)n][BF4]2 salts (M = NiII, PdII, and PtII; n = 4 or 6) affords di-radical tri-metallic complexes in a stairstep type arrangement ([FeMFe]2+, M = Ni, Pd, and Pt), with the central group 10 metal held in a MS4 square plane. These isostructural compounds have nearly identical ν(NO) stretching values, isomer shifts, and electrochemical properties, but vary in their magnetic properties. Despite the intramolecular Fe...Fe distances of ca. Å, antiferromagnetic coupling is observed between {Fe(NO)}7 units as established by magnetic susceptibility, EPR, and DFT studies. The superexchange interaction through the thiolate sulfur and central metal atoms is on the order of NiII < PdII ≪ PtII with exchange coupling constants (J) of −3, −23, and −124 cm−1, consistent with increased covalency of the M–S bonds (3d < 4d < 5d). This trend is reproduced by DFT calculations with molecular orbital analysis providing insight into the origin of the enhancement in the exchange interaction. Specifically, the magnitude of the exchange interaction correlates surprisingly well with the energy difference between the HOMO and HOMO−1 orbitals of the triplet states, which is reflected in the central metal's contribution to these orbitals. These results demonstrate the ability of sulfur-dense metallodithiolate ligands to engender strong magnetic communication by virtue of their enhanced covalency and polarizability. |
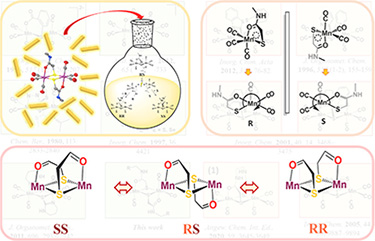 Chirality-Guided Isomerization of Mn2S2 Diamond Core Complexes: A Mechanistic Study Inorg. Chem. 2022, 61 (41), 16405-16413. DOI: 10.1021/acs.inorgchem.2c02460 |
Occasioned by the discovery of a ligand transfer from M(N2S2) to MnI in Mn(CO)5Br, the resulting H2N2S2 ligand-tethered dimanganese complex, (μ4-N,N′-ethylenebis(mercaptoacetamide))[Mn2(CO)6], was found to have myriad analogues of the type (μ-S–E)2[Mn2(CO)6], making up an under-studied class containing Mn2S2 rhombs. The attempt to synthesize a nontethered version resulted in a solid-state structure in an anti-conformation. However, a direct comparison of the Fourier-transform infrared spectra of the tethered versus nontethered complexes in combination with theoretical frequency calculation suggested the coexistence of syn- and anti-isomers and their interconversion in solution. Analysis of the syn- versus anti-version of the dimanganese components led to the understanding that whereas the anti-form exists as centrosymmetric RS isomers, the syn-form is restricted by C2 symmetry to be either RR or SS. Molecular scrambling experiments indicated monomeric, pentacoordinate, 16-e– (S–O)Mn(CO)3 intermediates with lifetimes sufficiently long to sample R and S monomers. Density functional theory analysis of the mechanistic pathway and a kinetic study corroborated that the proposed isomerization involves the cleavage and reformation of the dimeric structures. |
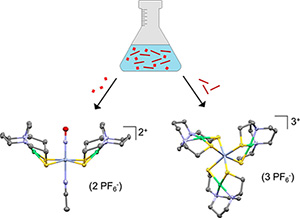 Structural Analysis of Heteropolymetallic Complexes: A Chiral Pinwheel {Cr[Ni(N2S2)]3}3+ and the {trans-(MeCN)Cr(NO)[Ni(N2S2)]2}2+ basket Polyhedron 2022, 224 (15), 116018. DOI: 10.1016/j.poly.2022.116018 |
The [Cr(NO)(MeCN)5]2+ synthon reacts with the Ni(N2S2) metallodithiolate ligand to yield co-crystallized products from which crystals suitable for SC-XRD could be extracted. The major product, {trans-(MeCN)Cr(NO)[Ni(N2S2)]2}2+, abbreviated [Cr(NO)Ni2]2+, is a symmetrical butterfly with upswept Ni(N2S2) wings flanking the trans-Cr(NO)(MeCN) backbone and designated {Cr(NO)}5 in Enemark-Feltham notation (Feltham and Enemark, 1981). The minor product, {Cr[Ni(N2S2)]3}3+, abbreviated [CrNi3]3+, is a pinwheel-type structure analogous to [RhNi3]3+, and both are comparable to tris-bipyridyl analogues. |
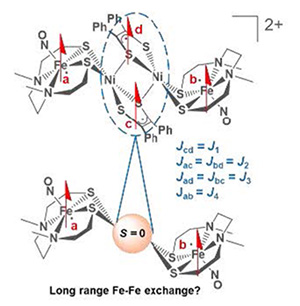 Cooperative Redox and Spin Activity from Three Redox Congeners of Sulfur-Bridged Iron Nitrosyl and Nickel Dithiolene Complexes PNAS 2022, 119 (25). DOI: 10.1073/pnas.2201240119 |
The synthesis of sulfur-bridged Fe–Ni heterobimetallics was inspired by Nature’s strategies to “trick” abundant first row transition metals into enabling 2-electron processes: redox-active ligands (including pendant iron–sulfur clusters) and proximal metals. Our design to have redox-active ligands on each metal, NO on iron and dithiolene on nickel, resulted in the observation of unexpectedly intricate physical properties. The metallodithiolate, (NO)Fe(N2S2), reacts with a labile ligand derivative of [NiII(S2C2Ph2)]0, NiDT, yielding the expected S-bridged neutral adduct, FeNi, containing a doublet {Fe(NO)}7. Good reversibility of two redox events of FeNi led to isolation of reduced and oxidized congeners. Characterization by various spectroscopies and single-crystal X-ray diffraction concluded that reduction of the FeNi parent yielded [FeNi]−, a rare example of a high-spin {Fe(NO)}8, described as linear FeII(NO–). Mössbauer data is diagnostic for the redox change at the {Fe(NO)}7/8 site. Oxidation of FeNi generated the 2[FeNi]+⇌[Fe2Ni2]2+ equilibrium in solution; crystallization yields only the [Fe2Ni2]2+ dimer, isolated as PF6− and BArF− salts. The monomer is a spin-coupled diradical between {Fe(NO)}7 and NiDT+, while dimerization couples the two NiDT+ via a Ni2S2 rhomb. Magnetic susceptibility studies on the dimer found a singlet ground state with a thermally accessible triplet excited state responsible for the magnetism at 300 K (χMT = 0.67 emu·K·mol−1, µeff = 2.31 µB), and detectable by parallel-mode EPR spectroscopy at 20 to 50 K. A theoretical model built on an H4 chain explains this unexpected low energy triplet state arising from a combination of anti- and ferromagnetic coupling of a four-radical molecular conglomerate. |
 Organometallic Chemistry Control of Hydrogenases contrubution in Enzymes for Solving Humankind's Problems Springer, Cham. 2021, 275-300. DOI: 10.1007/978-3-030-58315-6_10 |
This chapter provides an abbreviated overview of the discovery, exploration, and current knowledge of hydrogenase enzymes with particular regard to the mechanisms of electrocatalytic proton reduction in both the natural enzymes and synthetic analogues of the active sites. The timeline of key breakthroughs for defining the enzymes as well as in hydrogenase-inspired biomimetic research serves as a basis for the discussion. |
 Linear and Bent Nitric Oxide Ligand Binding in an Asymmetric Butterfly Complex: CoMoCo′ Inorg. Chem. 2021, 60 (21), 15975-15979 DOI: 10.1021/acs.inorgchem.1c00987 |
Two synthetic approaches to install metallodithiolate ligands on molybdenum centers using the synthons [Mo2(CH3CN)10]4+ and (N2S2)Co(NO) [N2S2 = N,N-bis(2-mercaptoethyl)-1,4-diazacycloheptane and NO = nitric oxide], or [Mo(NO)2(CH3CN)4]2+ (CH3CN = acetonitrile) and [(N2S2)Co]2 lead to a bis-nitrosylated, trimetallic dication, CoMoCo′. This unique asymmetric butterfly complex, with S = 1, has a bent NO within the small {Co(NO)}8 wing (denoted as Co), reflecting CoIII(NO–), and is S-bridged to a linear {Mo(NO)}6 diamagnetic unit. The latter is further S-bridged to a pentacoordinate (N2S2)CoIII(CH3CN) donor in the larger wing and is the origin of the two unpaired electrons, denoted as Co′. The asymmetry in Mo–Co distances, 3.33 Å in the Co wing and 2.73 Å in the Co′ wing, indicated a Mo–Co′ bonding interaction. The transfer of NO from (N2S2)Co(NO) in the former path is needed to cleave the strong quadruple bond in [Mo≣Mo]4+, with the energetic cost compensated for via a one-electron bond between Mo and Co′, as indicated by natural bonding orbital analysis. |
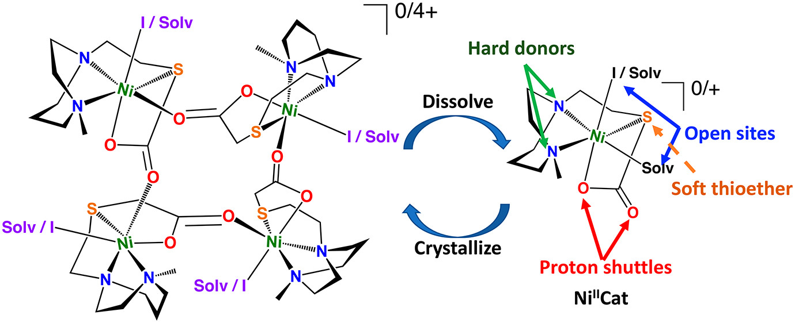 Self-Assembled Nickel-4 Supramolecular Squares and Assays for HER Electrocatalysts Derived Therefrom Inorg. Chem. 2021, 60 (10), 7051-7061. DOI: 10.1021/acs.inorgchem.0c03613 |
Solid-state structures find a self-assembled tetrameric nickel cage with carboxylate linkages, [Ni(N2S′O)I(CH3CN)]4 ([Ni-I]40), resulting from sulfur acetylation by sodium iodoacetate of an [NiN2S]22+ dimer in acetonitrile. Various synthetic routes to the tetramer, best described from XRD as a molecular square, were discovered to generate the hexacoordinate nickel units ligated by N2Sthioether, iodide, and two carboxylate oxygens, one of which is the bridge from the adjacent nickel unit in [Ni-I]40. Removal of the four iodides by silver ion precipitation yields an analogous species but with an additional vacant coordination site, [Ni-Solv]+, a cation but with coordinated solvent molecules. This also recrystallizes as the tetramer [Ni-Solv]44+. In solution, dissociation into the (presumed) monomer occurs, with coordinating solvents occupying the vacant site [Ni(N2S′O)I(solv)]0, ([Ni-I]0). Hydrodynamic radii determined from 1H DOSY NMR data suggest that monomeric units are present as well in CD2Cl2. Evans method magnetism values are consistent with triplet spin states in polar solvents; however, in CD2Cl2 solutions no paramagnetism is evident. The abilities of [Ni-I]40 and [Ni-Solv]44+ to serve as sources of electrocatalysts, or precatalysts, for the hydrogen evolution reaction (HER) were explored. Cyclic voltammetry responses and bulk coulometry with gas chromatographic analysis demonstrated that a stronger acid, trifluoroacetic acid, as a proton source resulted in H2 production from both electroprecatalysts; however, electrocatalysis developed primarily from uncharacterized deposits on the electrode. With acetic acid as a proton source, the major contribution to the HER is from homogeneous electrocatalysis. Overpotentials of 490 mV were obtained for both the solution-phase [Ni-I]0 and [Ni-Solv]+. While the electrocatalyst derived from [Ni-Solv]+ has a substantially higher TOF (102 s–1) than [Ni-I]0 (19 s–1), it has a shorter catalytically active lifespan (4 h) in comparison to [Ni-I]0 (>18 h). |